Thiếu hụt hoặc kháng vitamin D và giảm phosphat máu
Vijaya Sarathia, Melkunte Shanthaiah Dhananjayaa, Manjiri Karlekarb, Anurag Ranjan Lilab
a Khoa Nội tiết, Viện Khoa học Y khoa và Nghiên cứu Vydehi, Bengaluru 560066, Ấn Độ
b Khoa Nội tiết, Trường Y Seth G S và Bệnh viện King Edward, Mumbai 400012, Ấn Độ
Dịch và chú thích: Bs. Lê Đình Sáng
Vitamin D chủ yếu được tổng hợp ở da (cholecalciferol) thông qua phơi nắng trong khi một phần nhỏ được hấp thu từ thực phẩm (ergocalciferol). Vitamin D tiếp tục được chuyển hóa thành 25-hydroxyvitamin D và 1,25-dihydroxy vitamin D (calcitriol) lần lượt ở gan và thận. Calcitriol là dạng hoạt động trung gian tác dụng của vitamin D thông qua thụ thể vitamin D (VDR) có mặt khắp nơi. Khiếm khuyết ở bất kỳ mức độ nào trong con đường này đều dẫn đến còi xương do thiếu hoặc kháng vitamin D. Thiếu vitamin D do dinh dưỡng là nguyên nhân hàng đầu gây còi xương và loãng xương khắp thế giới và đáp ứng tốt với bổ sung vitamin D. Rối loạn di truyền chuyển hóa vitamin D (còi xương phụ thuộc vitamin D, VDDR) chiếm một tỷ lệ nhỏ trong các trường hợp còi xương/loãng xương do thiếu calci. Khiếm khuyết hydroxyl hóa 1α vitamin D, hydroxyl hóa 25 vitamin D và thụ thể vitamin D lần lượt gây ra VDDR1A, VDDR1B và VDDR2A, trong khi gắn kết khiếm khuyết của vitamin D với phần tử đáp ứng vitamin D do biểu hiện quá mức của heterogeneous nuclear ribonucleoprotein và chuyển hóa vitamin D tăng tốc lần lượt gây ra VDDR2B và VDDR3. Hấp thu calci từ thức ăn suy giảm và thiếu hụt calci làm tăng hormone tuyến cận giáp trong các rối loạn này dẫn đến tăng bài tiết phosphat niệu và giảm phosphat máu. Giảm phosphat máu là đặc điểm phổ biến của tất cả các rối loạn này, mặc dù không phải là dấu hiệu bệnh và dẫn đến giảm khoáng hóa xương và bệnh cơ. Cải thiện tình trạng giảm phosphat máu là một trong những dấu hiệu sớm nhất của đáp ứng với bổ sung vitamin D trong còi xương/loãng xương do dinh dưỡng và thiếu đáp ứng như vậy nên thúc đẩy đánh giá các dạng di truyền của còi xương/loãng xương.
I. GIỚI THIỆU
Vitamin D chủ yếu được sản xuất ở da thông qua phơi nắng. Khi tiếp xúc với tia cực tím B (∼290-315 nm), 7-dehydrocholesterol (tiền vitamin D3) trong da được chuyển đổi thành previtamin D3 và sau đó đồng phân hóa nhiệt thành vitamin D3 để đi vào tuần hoàn. Vitamin D3 liên kết với protein gắn vitamin D trong tuần hoàn và được vận chuyển đến gan, nơi nó được chuyển đổi thành 25-hydroxyvitamin D (25OHD). Hydroxyl hóa ở vị trí 25 của vitamin D chủ yếu được thực hiện bởi 25-hydroxylase vi thể được mã hóa chủ yếu bởi CYP2R1. 25OHD là dạng vitamin D lưu hành phổ biến nhất và cũng là chỉ số đáng tin cậy nhất về tình trạng vitamin D. 25OHD sau đó được chuyển đổi trong thận thành 1,25(OH)2 vitamin D [1,25(OH)2D] (dạng hoạt động nhất của vitamin D) bởi 1α-hydroxylase được mã hóa bởi CYP27B1. Hydroxyl hóa ở vị trí 24 của 25OHD và 1,25(OH)2D thành 24,25(OH)2D và 1,24,25(OH)3D tương ứng là con đường chính để bất hoạt vitamin D và được trung gian bởi 24-hydroxylase được mã hóa bởi CYP24A1. CYP3A4 đóng vai trò phụ trong việc bất hoạt vitamin D, chuyển đổi 25OHD và 1,25(OH)2D thành 4β,25(OH)2D và 1,23,25(OH)3D tương ứng.
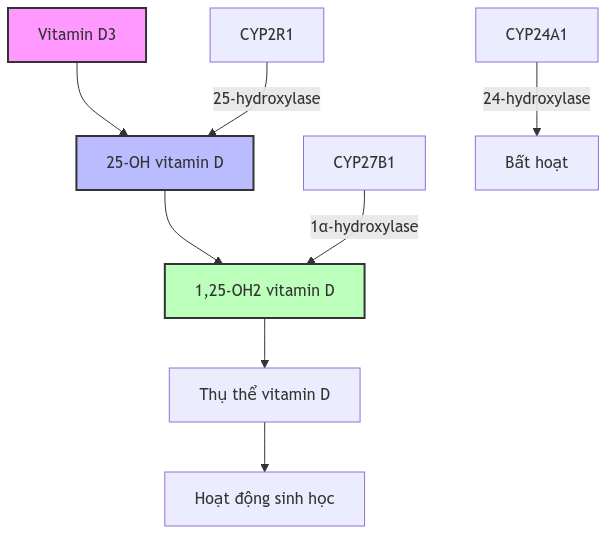
Hình 1. Sơ đồ quá trình chuyển hóa Vitamin D
II. VAI TRÒ CỦA VITAMIN D TRONG CHUYỂN HÓA PHOSPHAT
Phosphorus từ thức ăn được hấp thu ở ruột non, chủ yếu ở tá tràng và hỗng tràng. Hấp thu phosphat ở ruột được thực hiện bởi ít nhất hai con đường riêng biệt: qua tế bào và cạnh tế bào. Vận chuyển phosphat thụ động cạnh tế bào thông qua Na+/H+ exchanger 3 (NHE3, SLC9A3) chiếm phần lớn (65-80%) hấp thu phosphat ở ruột, trong khi hấp thu phosphat qua tế bào chiếm phần còn lại và được trung gian bởi chất vận chuyển phosphat phụ thuộc natri type II 2b (NaPi-IIb, SLC34A2). Cơ chế sau được điều hòa bởi vitamin D, chiếm khoảng 30% hấp thu phosphat phụ thuộc vitamin D.
III. VAI TRÒ CỦA VITAMIN D TRONG CHUYỂN HÓA PHOSPHAT (tiếp)
Nồng độ phosphorus thấp trong máu dẫn đến tăng tổng hợp 1,25 dihydroxy vitamin D, độc lập với nồng độ PTH, thông qua điều hòa tăng phiên mã của gen 1α-hydroxylase. Có vẻ như tác động kích thích của phosphorus lên sản xuất 1,25(OH)2D phụ thuộc vào trục GH/IGF-1 nguyên vẹn. Thú vị là sự điều hòa calci-phosphat bởi vitamin D chủ yếu thông qua các tác động qua gen. Tuy nhiên, có một số bằng chứng ủng hộ tác động không qua gen của nó trong hấp thu phosphorus ở ruột thông qua protein disulfide isomerase họ A thành viên 3 (PDIA3).
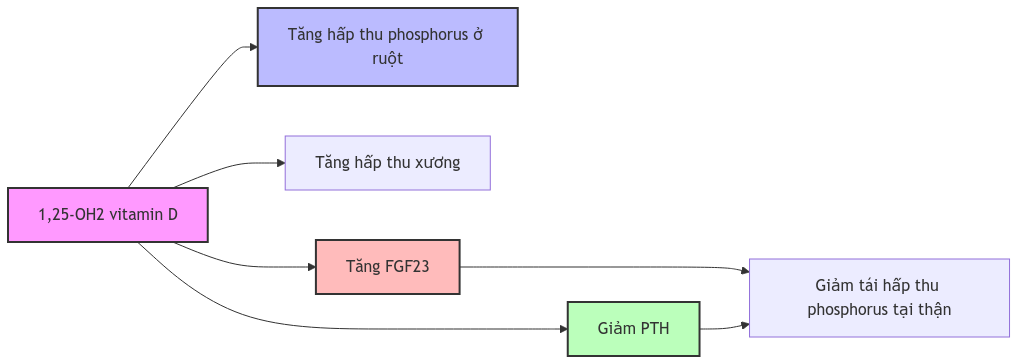
Hình 2. Cơ chế điều hòa phosphorus bởi Vitamin D
Vitamin D dường như không có vai trò trực tiếp trong điều hòa xử lý phosphorus ở thận. Tuy nhiên, nó có thể có tác động gián tiếp thông qua tăng FGF23 có thể làm giảm tái hấp thu phosphat, điều này có thể bù trừ nhẹ tác động tích cực của vitamin D lên nồng độ phosphorus máu. 1,25(OH)2D cũng có thể đóng vai trò trong tái hấp thu phosphat từ xương. 1,25(OH)2D chủ yếu tác động lên nguyên bào xương, sau đó kích thích hủy cốt bào để gây hấp thu xương. Hoạt động hủy cốt bào tăng này cũng được quan sát thấy ở chuột cắt tuyến giáp-cận giáp, cho thấy vai trò độc lập với PTH của 1,25(OH)2D trong hấp thu xương.
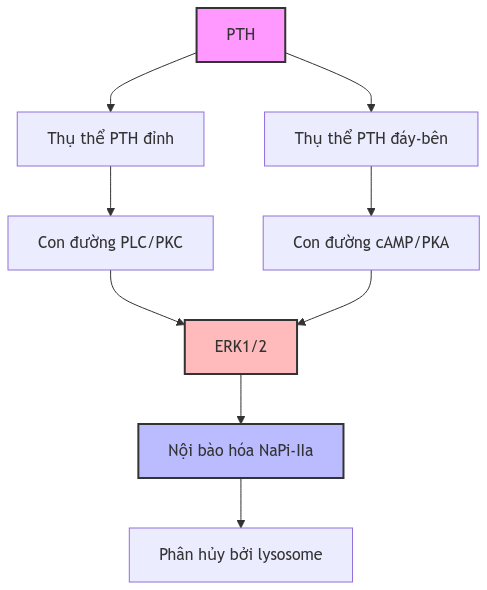
Hình 3. Cơ chế điều hòa phosphorus bởi PTH tại ống thận
Chú thích: PTH: Hormone tuyến cận giáp; PLC: Phospholipase C; PKC: Protein kinase C; cAMP: Cyclic adenosine monophosphate; PKA: Protein kinase A; ERK1/2: Extracellular signal-regulated kinases 1/2; NaPi-IIa: Na+-dependent phosphate cotransporter IIa
IV. THIẾU HỤT VITAMIN D VÀ CÁC RỐI LOẠN LIÊN QUAN
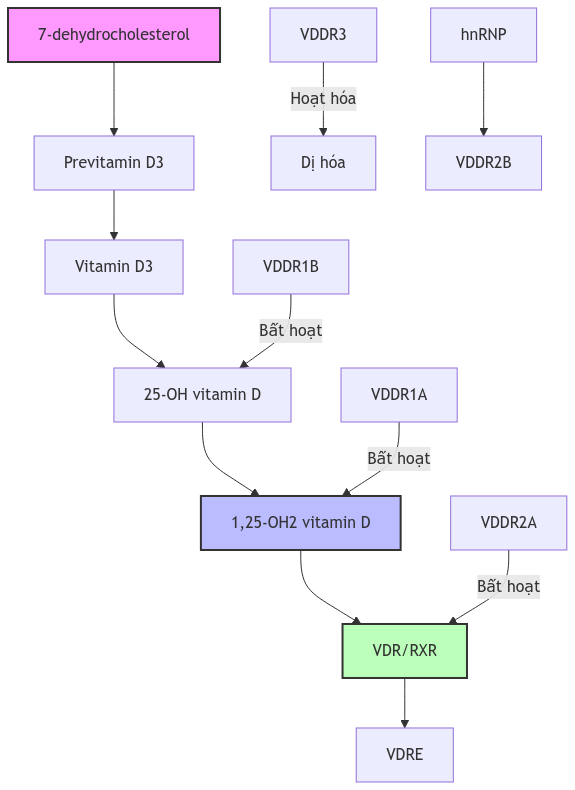
Hình 4. Con đường chuyển hóa vitamin D và các khiếm khuyết liên quan
Chú thích: VDDR: Còi xương phụ thuộc vitamin D; VDR: Thụ thể vitamin D; RXR: Thụ thể retinoid X; VDRE: Yếu tố đáp ứng vitamin D; hnRNP: Heterogeneous nuclear ribonucleoprotein
4.1. Thiếu vitamin D
Tỷ lệ trung bình toàn cầu (95% CI) của nồng độ 25OHD huyết thanh < 30, < 50, và < 75 nmol/L lần lượt là 15,7% (13,7-17,8), 47,9% (44,9-50,9), và 76,6% (74,0-79,1). Mặc dù có sự giảm nhẹ tỷ lệ thiếu vitamin D trong giai đoạn 2011-2022 so với 2000-2012, tỷ lệ vẫn còn cao. Tỷ lệ thiếu vitamin D còn cao hơn trong mùa đông-xuân, ở phụ nữ, và người sống ở vùng cao, các nước thu nhập trung bình thấp và các nước Đông-Địa Trung Hải.
4.2. Mối liên quan giữa phosphat huyết thanh và 25OHD
Một nghiên cứu bao gồm trẻ em từ Vương quốc Anh cho thấy mối liên quan đáng kể giữa 25OHD thấp với phosphat huyết thanh (p = 0,004). Ngược lại, một nghiên cứu lớn trên bệnh nhân ngoại trú (n = 23.134) từ Đức (r = 0,01, p = 0,529) và một nghiên cứu dựa trên hồ sơ bệnh viện từ Thổ Nhĩ Kỳ (r = 0,016, p = 0,62) không báo cáo mối tương quan đáng kể giữa nồng độ phosphorus và 25OHD trong máu.
IV. THIẾU HỤT VITAMIN D VÀ CÁC RỐI LOẠN LIÊN QUAN (tiếp)
4.3. Vai trò của giảm phosphat máu trong còi xương
Còi xương/loãng xương do dinh dưỡng phát triển dần dần trong vài tháng đến vài năm. Giai đoạn I đặc trưng bởi giảm calci máu tạm thời kèm theo tăng nhẹ phosphatase kiềm huyết thanh. Phần lớn giai đoạn này không được nhận biết và tiến triển sang giai đoạn II – giai đoạn phổ biến nhất khi bệnh nhân đến khám. Giai đoạn II và III đặc trưng bởi giảm phosphat máu đóng vai trò chính trong quá trình khoáng hóa khiếm khuyết trong còi xương do dinh dưỡng.
Vai trò quan trọng hơn của phosphorus trong khoáng hóa khiếm khuyết được chứng minh bởi sự xuất hiện của nó ngay cả ở bệnh nhân có rối loạn giảm phosphat (còi xương giảm phosphat liên kết X). Thú vị là, thiếu hụt calci trong giai đoạn sơ sinh và sơ nhi thường không biểu hiện thành còi xương vì cần phải có tình trạng giảm phosphat máu kéo dài do cường cận giáp thứ phát trước khi các dấu hiệu X-quang của còi xương trở nên rõ ràng. Khoáng hóa xương ở sụn tăng trưởng được khởi đầu bởi một chuỗi sự kiện được kích hoạt bởi quá trình chết theo chương trình của tế bào sụn phì đại. Cần có phosphat để thực hiện quá trình chết theo chương trình của tế bào sụn phì đại qua trung gian caspase 9; do đó, giảm phosphat máu là cơ sở của cả còi xương do giảm phosphat và thiếu calci.
Giảm phosphat máu trong thiếu vitamin D có thể nặng hơn so với cường cận giáp nguyên phát, vì giảm hấp thu phosphorus ở ruột cũng góp phần gây giảm phosphat máu trong thiếu vitamin D. Đáng chú ý, phosphorus máu có thể bình thường ở 1/4 đến 1/3 trẻ em bị còi xương do dinh dưỡng từ các quốc gia khác nhau. Thú vị là, giảm phosphat máu còn ít rõ rệt hơn ở trẻ em bị còi xương do thiếu calci từ Nigeria. Một nhóm người thiếu vitamin D có thể có tăng phosphat máu, chủ yếu liên quan đến suy thận. Tuy nhiên, khi tăng phosphat máu được ghi nhận ở những người có chức năng thận bình thường, nó có thể do kháng tác dụng của PTH liên quan đến thiếu vitamin D (giả suy cận giáp type 2).
|
Bảng 1. Nguyên nhân phổ biến của thiếu vitamin D Yếu tố bên ngoài:
Yếu tố bên trong:
Các tình trạng gây tăng dị hóa hoặc bài tiết:
|
Bảng 2. Các giai đoạn thiếu vitamin D và đặc điểm sinh hóa
| Giai đoạn | 25OHD | Calci | Phosphorus | ALP | PTH |
|---|---|---|---|---|---|
| Giai đoạn 1 | ↓ | ↓ | ↔ | ↔↑ | ↔↑ |
| Giai đoạn 2 | ↓↓ | ↔ | ↓ | ↑↑ | ↑↑ |
| Giai đoạn 3 | ↓↓↓ | ↓↓ | ↓↓ | ↑↑↑ | ↑↑↑ |
Chú thích các ký hiệu trong bảng: ↑: tăng; ↓: giảm; ↔: không thay đổi; ALP: Alkaline phosphatase
PTH: Hormone tuyến cận giáp; 25OHD: 25-hydroxyvitamin D
4.4. Giả suy cận giáp type 2 và thiếu vitamin D
Có nhiều trường hợp được công bố về thiếu vitamin D bắt chước hoặc bị chẩn đoán nhầm là giả suy cận giáp type 2. Hầu hết các trường hợp này có giảm calci máu với một hoặc nhiều biểu hiện giảm calci máu, tăng phosphat và PTH huyết thanh kèm thiếu vitamin D nặng. Thú vị là, điều trị vitamin D đã bình thường hóa calci máu và giảm cả phosphorus và PTH huyết thanh ở đa số trường hợp. Do đó, nên thử điều trị vitamin D ở những bệnh nhân thiếu vitamin D nặng nhưng có đặc điểm sinh hóa của giả suy cận giáp.
4.5. Giảm phosphat máu và yếu cơ
Ngoại trừ loãng xương giảm phosphat liên kết X, yếu cơ là đặc điểm phổ biến của hầu hết các dạng loãng xương bao gồm loãng xương do dinh dưỡng và phổ biến hơn so với cường cận giáp nguyên phát. Thật vậy, loãng xương do dinh dưỡng vẫn là nguyên nhân phổ biến gây bệnh cơ gần ở nhiều quốc gia. Tất cả các tế bào sống đều sử dụng phosphat như một cơ chất quan trọng cho chuyển hóa. Trong cơ xương, phosphat được dự trữ dưới dạng phosphorus hữu cơ, chủ yếu dưới dạng adenosine triphosphate (ATP) và phosphoryl creatinine. Các chất vận chuyển Pit1 và Pit2 kiểm soát nồng độ phosphorus vô cơ tự do nội bào (Pi), khoảng 1-2 mg/dL (3-5 mmol) trong tế bào cơ. Lượng phosphorus này trong tế bào cơ cần thiết để duy trì dự trữ creatinine phosphate và hiệu quả của ATP như một nguồn năng lượng cho hoạt động cơ học của cơ. Nồng độ Pi máu thấp có thể dẫn đến suy giảm sản xuất ATP cơ, điều này có thể giải thích một số yếu cơ và bệnh cơ quan sát thấy ở bệnh nhân còi xương giảm phosphat.
Đáng chú ý, một số bệnh nhân loãng xương do dinh dưỡng có phosphorus máu bình thường. Điều này có thể gợi ý ngưỡng phosphat huyết thanh để gây loãng xương do dinh dưỡng khác nhau giữa các cá nhân hoặc vai trò của các yếu tố khác ngoài phosphorus máu như tác động trực tiếp của thiếu vitamin D và PTH. PTH tăng cao làm suy giảm hoạt động của creatinine phosphokinase ty thể và myofibril và ATPase dẫn đến suy giảm sản xuất, vận chuyển và sử dụng năng lượng.
Nhiều nghiên cứu đã báo cáo mối liên quan giữa PTH tăng cao, độc lập với tình trạng vitamin D, với tác động bất lợi lên sức mạnh cơ và sự ổn định tư thế; tuy nhiên, các phân tích xem mối liên quan này có độc lập với phosphorus máu hay không thì chưa có. Vai trò trực tiếp của vitamin D trong bệnh cơ cũng mới được đề xuất gần đây vì thiếu vitamin D có thể dẫn đến stress oxy hóa và teo cơ.
4.6. Đáp ứng phosphorus máu với bổ sung vitamin D trong thiếu vitamin D
Điều trị với vitamin D, dù là liều hàng ngày hay liều tấn công, làm tăng phosphorus máu sau 4-8 ngày với đỉnh nồng độ giữa 2-4 tuần và đồng thời tăng TMP/GFR và giảm PTH. Thú vị là, sự tăng nồng độ phosphorus máu sau bổ sung vitamin D thường quá mức và có thể kéo dài đến 4-6 tháng.
Tăng phosphorus máu được coi là đáp ứng sinh hóa sớm nhất với liệu pháp tấn công. Một số tác giả gợi ý rằng việc không bình thường hóa phosphorus máu sau một tuần bổ sung vitamin D có thể cho thấy không đáp ứng với vitamin D. Tuy nhiên, trong một nghiên cứu gần đây từ Ấn Độ, liệu pháp tấn công không bình thường hóa được phosphorus máu ở 17% và 9% trẻ em còi xương sau 3 và 6 tuần điều trị, nhưng đã bình thường hóa phosphorus máu ở tất cả bệnh nhân sau 3 tháng. Ngoài ra, nồng độ phosphorus máu đạt đỉnh muộn hơn, tức là ở tháng 3-6 và duy trì đến tháng 12. Trong một nghiên cứu khác từ Qatar, liệu pháp tấn công đã bình thường hóa phosphorus máu ở tất cả trẻ em còi xương sau 1 tháng. Thay đổi phosphorus máu sau liệu pháp tấn công có tương quan dương với thay đổi calci máu (r = 0,256) và tương quan âm với thay đổi phosphatase kiềm (r = 0,276) và PTH (r = 0,366). Đáng ngạc nhiên, thay đổi phosphorus máu không tương quan với thay đổi 25OHD máu (r = 0,067).
V. CÒI XƯƠNG PHỤ THUỘC VITAMIN D TYPE 1A (VDDR1A)
Tác dụng sinh học của vitamin D chủ yếu được trung gian bởi 1,25(OH)2D (calcitriol). Chuyển đổi 25OHD thành 1,25(OH)2D được xúc tác bởi 1α-hydroxylase (CYP27B1) ở thận. Đột biến hai alen trong gen mã hóa 1α-hydroxylase (CYP27B1, định vị trên nhiễm sắc thể 12q13) gây thiếu hụt 1,25(OH)2D và dẫn đến VDDR1A (giả thiếu vitamin D). VDDR1A được di truyền theo kiểu lặn trên nhiễm sắc thể thường. Các triệu chứng thường gặp khi khởi phát bao gồm dị dạng, chậm phát triển vận động, giảm trương lực cơ, chậm phát triển hoặc co giật do giảm calci máu.
Kết quả xét nghiệm điển hình bao gồm giảm calci máu, giảm phosphat máu, tăng phosphatase kiềm (ALP) và PTH huyết thanh, và nồng độ 1,25(OH)2D thấp hoặc thấp-bình thường. Trong một đánh giá tổng quan hệ thống gần đây của nhóm chúng tôi, nồng độ phosphorus máu trung vị ở bệnh nhân VDDR1A là 3,8 (2,4-4,99) mg/dl, với tình trạng giảm phosphat máu chỉ được quan sát thấy ở khoảng ba phần tư số bệnh nhân. Tuy nhiên, chưa rõ liệu sự hiện diện của giảm phosphat máu có cho thấy bệnh nặng hơn hay không. Trong đánh giá tổng quan hệ thống, nồng độ phosphorus máu không khác biệt giữa các bệnh nhân VDDR1A có đột biến cắt ngắn và không cắt ngắn. Tuy nhiên, một nghiên cứu sau đó báo cáo giảm phosphat máu nhẹ hơn [-2,4 (-3,5 đến -1,5) vs. -3,6 (-7,3 đến -1,1) ở bệnh nhân có bệnh nhẹ hơn (p.Ala129Thr), mặc dù không có ý nghĩa thống kê.
VDDR1A là một tình trạng được biết đến với việc thường bị chẩn đoán sai ban đầu, phổ biến nhất là còi xương do dinh dưỡng nhưng cũng có thể là nhiễm toan ống thận xa hoặc còi xương giảm phosphat. Gần đây, cũng có báo cáo về chẩn đoán nhầm VDDR1A thành cường cận giáp nguyên phát có calci máu bình thường. Thú vị hơn, mặc dù giảm phosphat máu là một phát hiện thường gặp, phosphorus máu đôi khi có thể tăng cao dẫn đến chẩn đoán nhầm là giả suy cận giáp.
Nồng độ 1,25(OH)2D thấp ở bệnh nhân có loãng xương/còi xương do thiếu calci (tăng PTH) không đáp ứng với liều vitamin D chuẩn, đặc biệt khi có nồng độ 25OHD bình thường-cao, là đặc trưng của VDDR1A. Tuy nhiên, ở một số bệnh nhân, 1,25(OH)2D có thể bình thường hoặc thậm chí tăng nhẹ. Quan sát sau thường được thấy nhiều hơn ở bệnh nhân có 25OHD cao-bình thường đến tăng cao. Trong những trường hợp như vậy, tỷ lệ 1,25(OH)2D/25OHD thấp hơn (0,31) có thể tăng độ nhạy chẩn đoán. Tuy nhiên, nồng độ 1,25(OH)2D bình thường cũng được ghi nhận ở một số bệnh nhân VDDR1A có thiếu vitamin D, điều này cảnh báo cần giải thích cẩn thận và xem xét xét nghiệm gen để chẩn đoán chính xác.
Liều calcitriol thông thường là khoảng 45 [30-60] ng/kg/ngày. Thời gian chậm khoảng 5 tháng và 1 năm để bình thường hóa calci máu và phosphatase kiềm được quan sát trong nghiên cứu gần đây của Pháp, trong khi một nghiên cứu khác từ Trung Quốc cho thấy ALP và PTH trở về bình thường sau 3 và 6 tháng điều trị. Có hạn chế các nghiên cứu nhấn mạnh về việc bình thường hóa phosphorus máu với điều trị trong VDDR1A. Phosphorus máu là 0,87 ± 0,23 mmol/L khi chẩn đoán (2,1 ± 0,8 tuổi) và tăng sau điều trị lên 1,29 ± 0,30 tại lần khám cuối (8,2 ± 4,7 tuổi). Đáng chú ý, phosphorus máu vẫn thấp ở khoảng một nửa số bệnh nhân và liên quan đến tăng PTH dai dẳng cho thấy điều trị chưa đủ ở những bệnh nhân này.
VI. CÒI XƯƠNG PHỤ THUỘC VITAMIN D TYPE 1B (VDDR1B)
Vitamin D có thời gian bán hủy ngắn (<1-2 ngày) trong khi 25OHD (calcifediol) có thời gian bán hủy dài hơn (2-3 tuần) khiến nó trở thành dạng vitamin D lưu hành phổ biến nhất. Hydroxyl hóa vị trí 25 của vitamin D chủ yếu được trung gian bởi CYP2R1 và các đột biến bất hoạt trong CYP2R1 làm giảm chức năng 25-hydroxylase là nguyên nhân chính của VDDR1B. Đây là một rối loạn tương đối hiếm gặp với chỉ khoảng 50 ca được báo cáo cho đến nay. Tuy nhiên, tình trạng này có thể bị chẩn đoán thiếu vì nó bắt chước thiếu vitamin D do dinh dưỡng (giả thiếu vitamin D do dinh dưỡng). Đây là một tình trạng bán trội trên nhiễm sắc thể thường với bệnh biểu hiện cả ở các biến thể một alen và hai alen, mặc dù biểu hiện nhẹ hơn ở những người có biến thể một alen.
Đánh giá sinh hóa giống với thiếu vitamin D đặc trưng bởi giảm 25OHD máu, giảm calci máu, giảm phosphat máu và tăng PTH. Tuy nhiên, giảm đáp ứng với liều vitamin D chuẩn phân biệt tình trạng này với thiếu vitamin D. Cần có chỉ số nghi ngờ cao để chẩn đoán tình trạng này. Những bệnh nhân cần điều trị liên tục với liều cao vitamin D để duy trì nồng độ 25OHD nên được xem xét xét nghiệm gen CYP2R1.
VDDR1B có thể được điều trị theo nhiều cách. Liều sinh lý của calcitriol hoặc liều dược lý của ergocalciferol hoặc cholecalciferol kết hợp với bổ sung calci là một chiến lược điển hình. Calcifediol có sẵn ở một số quốc gia và là phương pháp cải tiến để điều trị VDDR1B vì nó tránh được khiếm khuyết hydroxyl hóa 25.
VII. CÒI XƯƠNG PHỤ THUỘC VITAMIN D TYPE 2A (VDDR2A)
VDDR2A, còn gọi là còi xương kháng vitamin D di truyền (HVDRR), là một rối loạn hiếm gặp gây ra bởi sự kháng của cơ quan đích với tác dụng của vitamin D và được di truyền theo kiểu lặn trên nhiễm sắc thể thường. VDDR2A thường do các biến thể bệnh lý hai alen trong gen VDR mã hóa thụ thể vitamin D (VDR) và nằm trên nhiễm sắc thể 12q13.11. Bệnh nhân VDDR2A thường biểu hiện với còi xương do thiếu calci từ sớm với khởi phát bệnh ở 19 [12-24] tháng tuổi. Khoảng một nửa số bệnh nhân có thể có chậm phát triển khi biểu hiện trong khi bất thường răng (giảm sản men) và viêm phổi tái phát có thể xuất hiện ở 10-15% bệnh nhân. Rụng tóc là đặc điểm đặc trưng của HVDDR và được quan sát thấy ở bốn phần năm số bệnh nhân. Rụng tóc từng được coi là dấu hiệu của đáp ứng kém với calci và calcitriol đường uống. Tuy nhiên, trong một đánh giá gần đây, chúng tôi đã báo cáo không có mối liên quan giữa rụng tóc và đáp ứng với điều trị.
Đặc điểm sinh hóa điển hình của HVDDR bao gồm giảm calci máu, giảm phosphat máu, tăng ALP, PTH cao, nồng độ 25OHD bình thường/thấp và tăng 1,25(OH)2D. Thông số sau phân biệt nó với VDDR1A. Khi không có rụng tóc, VDDR2A rất giống với còi xương do thiếu calci phổ biến ở một số nước kém phát triển và đang phát triển. Tương tự như các dạng VDDR đã thảo luận ở trên, giảm phosphat máu có thể không xuất hiện ở một tỷ lệ bệnh nhân. Trong một đánh giá tổng quan hệ thống gần đây, phosphorus máu ở bệnh nhân VDDR2A là 03 (2,4-3,6) mg/dl với giảm phosphat máu chỉ xuất hiện ở khoảng 58% bệnh nhân. Nồng độ phosphorus máu không khác biệt giữa bệnh nhân VDDR2A có và không có rụng tóc [3,3(2,5-3,8) vs. 2,7 (2,3-3,8) mg/dl] hoặc giữa các bệnh nhân có đột biến cắt ngắn vùng liên kết với phối tử [2,9 (2,5-3,4) mg/dl], không cắt ngắn vùng liên kết với phối tử [3,2 (2,5-3,7) mg/dl], cắt ngắn vùng liên kết DNA [2,7 (2,2-3,5) mg/dl] và không cắt ngắn vùng liên kết DNA [3,3 (2,5-3,9) mg/dl].
Phác đồ điều trị thông thường là liều cao calci và calcitriol đường uống. Trong các trường hợp kháng trị, truyền calci tĩnh mạch trong vài tuần để đạt được khỏi bệnh về mặt X-quang rất hữu ích. Có hạn chế dữ liệu về đáp ứng phosphorus máu với điều trị trong VDDR2A. Nồng độ phosphorus máu ở bệnh nhân VDDR2A có và không có tuân thủ điều trị là tương đương, mặc dù PTH cao hơn đáng kể ở những bệnh nhân tuân thủ kém.
VIII. CÒI XƯƠNG PHỤ THUỘC VITAMIN D TYPE 2B (VDDR2B)
Đây là một tình trạng ít được xác định rõ, bắt chước các đặc điểm lâm sàng và sinh hóa của VDDR type 2 nhưng không có bất thường nhận biết được trong gen VDR. Năm 1993, một bệnh nhân có đặc điểm lâm sàng và sinh hóa điển hình (bao gồm giảm phosphat máu) được báo cáo không có bất thường phân tử trong gen VDR. VDR từ bệnh nhân này có thể liên kết với calcitriol nhưng không thể định vị nhân và do đó, không có đáp ứng chức năng với calcitriol.
Trong các nghiên cứu biểu hiện, VDR của bệnh nhân cho thấy hoạt hóa chuyển mã bình thường khi có mặt calcitriol, gợi ý một VDR hoạt động bình thường. Sau đó, một nghiên cứu mở rộng của nhóm đã chứng minh sự can thiệp bởi heterogeneous nuclear ribonucleoprotein (hnRNP) biểu hiện quá mức theo cấu trúc với sự liên kết của dimer VDR-thụ thể retinoid X (RXR) hoạt động bình thường với yếu tố đáp ứng vitamin D (VDRE). Giraldo và cộng sự đã báo cáo hơn 200 trẻ em bị còi xương có đặc điểm sinh hóa của VDDR2 nhưng trình tự VDR bình thường được chẩn đoán là VDDR2B. Những trẻ còi xương này có nồng độ phosphorus cao hơn trẻ bình thường mặc dù có PTH cao hơn và liên quan đến bài tiết phosphorus niệu thấp hơn, trong khi điều trị với liều cao calcitriol và calci phosphat không dẫn đến thay đổi nồng độ phosphorus máu nhưng làm tăng bài tiết phosphorus niệu. Các đặc điểm chuyển hóa phosphorus ở những trẻ này rất thú vị và thiếu các nghiên cứu tiếp theo để xác nhận lại những phát hiện này.
IX. CÒI XƯƠNG PHỤ THUỘC VITAMIN D TYPE 3 (VDDR3)
VDDR3 là một tình trạng trội trên nhiễm sắc thể thường trong đó thiếu vitamin D được gây ra bởi chuyển hóa vitamin D tăng tốc. Nó được gây ra do đột biến tăng chức năng trong CYP3A4, mã hóa một enzyme P450. Đây là một tình trạng rất hiếm với chỉ ba ca được báo cáo cho đến nay. Thú vị là, cả ba bệnh nhân đều từ các gia đình không có quan hệ nhưng có cùng một đột biến (c.902 T > C, p.Iso301Thr) ở trạng thái đồng hợp tử. Đáng chú ý, protein đột biến với biến thể này không có tác động lên sự dị hóa của các cơ chất khác. Tình trạng này đặc trưng bởi còi xương khởi phát sớm, giảm nồng độ calci, phosphorus, 25OHD, 1,25(OH)2D trong máu và đáp ứng không đầy đủ với cả 25OHD và calcitriol.
Tăng cả 1,25(OH)2D và 25OHD vào ngày 3 sau khi dùng vitamin D (150.000 IU) trong VDDR3 tương đương với trẻ em bị còi xương do dinh dưỡng nhưng nồng độ trở nên thấp hơn đáng kể sau 7 ngày. Bệnh có thể được điều trị với liều siêu sinh lý của vitamin D (10.000-50.000 IU mỗi ngày) hoặc calcitriol. Bình thường hóa phosphorus máu được báo cáo sau điều trị đầy đủ.
Bảng 3. Các đặc điểm chính của các loại còi xương phụ thuộc vitamin D (VDDR)
| Loại | Nguyên nhân | Gen | Đặc điểm nổi bật | Xét nghiệm đặc trưng | Điều trị |
|---|---|---|---|---|---|
| VDDR1A | Khiếm khuyết 1α-hydroxylase | CYP27B1 | – Khởi phát sớm
– Yếu cơ – Co giật giảm calci |
– 1,25(OH)2D thấp
– 25OHD bình thường/cao |
Calcitriol |
| VDDR1B | Khiếm khuyết 25-hydroxylase | CYP2R1 | – Bắt chước thiếu vitamin D
– Kháng vitamin D liều chuẩn |
– 25OHD thấp
– Kháng vitamin D |
Calcifediol hoặc Calcitriol |
| VDDR2A | Đột biến thụ thể vitamin D | VDR | – Rụng tóc 80% ca
– Kháng calcitriol |
– 1,25(OH)2D cao
– PTH cao |
Calci liều cao
± Calcitriol |
| VDDR2B | Kháng VDR không đột biến | Không rõ | – Giống VDDR2A
– Không đột biến VDR |
– Tương tự VDDR2A
– VDR bình thường |
Tương tự VDDR2A |
| VDDR3 | Tăng dị hóa vitamin D | CYP3A4 | – Chuyển hóa vitamin D nhanh | – 25OHD và 1,25(OH)2D thấp | Vitamin D liều cao
hoặc Calcitriol |
Bảng 4. Thay đổi các chỉ số sinh hóa theo thời gian trong điều trị VDDR
| Thời điểm | Phosphorus máu | Calci máu | PTH | ALP |
|---|---|---|---|---|
| Khởi đầu | ↓↓ | ↓ | ↑↑ | ↑↑↑ |
| 1-2 tuần | ↑ | ↔/↑ | ↓ | ↔ |
| 1 tháng | ↑↑ | ↑ | ↓↓ | ↓ |
| 3-6 tháng | Bình thường | Bình thường | Bình thường | ↓↓ |
| > 6 tháng | Bình thường | Bình thường | Bình thường | Bình thường |
Chú thích: ↑: tăng nhẹ; ↑↑: tăng vừa; ↑↑↑: tăng nhiều; ↓: giảm nhẹ; ↓↓: giảm vừa; ↔: không thay đổi
Lưu ý quan trọng:
X. TÓM TẮT
Tóm lại, giảm phosphat máu là một đặc điểm phổ biến của thiếu vitamin D, kháng vitamin D và các rối loạn liên quan. Giảm phosphat máu đóng vai trò quan trọng trong khoáng hóa khiếm khuyết trong tất cả các rối loạn thiếu calci này. Tuy nhiên, phosphorus máu thường bình thường hoặc thậm chí hiếm khi tăng cao dẫn đến chẩn đoán nhầm là giả suy cận giáp. Điều trị đầy đủ và giảm PTH sẽ điều chỉnh tình trạng giảm phosphat máu trong tất cả các rối loạn thiếu calci. Điều chỉnh giảm phosphat máu là một trong những chỉ báo sinh hóa sớm nhất và đáng tin cậy về đáp ứng với điều trị. Tuy nhiên, việc nhấn mạnh về phosphorus máu trong chẩn đoán và theo dõi các rối loạn thiếu calci còn hạn chế và cần thêm các nghiên cứu tập trung vào cân bằng nội môi phosphorus trong các rối loạn này.
CHƯƠNG TRÌNH NGHIÊN CỨU
ĐIỂM THỰC HÀNH
TÀI LIỆU THAM KHẢO
Hơn 1 tháng mang cán thìa trong phế quản: Cuộc “giải cứu” ngay trong đêm
Nội soi siêu âm (EUS) – Bước tiến hiện đại trong chẩn đoán và điều trị bệnh lý tiêu hóa
Bệnh viện Hữu nghị đa khoa Nghệ An tổ chức hội thảo với chuyên đề “Tăng huyết áp và nguy cơ tim mạch”
Khép lại 23 năm sống chung với đường rò bàng quang – âm đạo bằng phẫu thuật nội soi chuyên sâu

https://ejournal.poltekkes-pontianak.ac.id/index.php/JKK
https://astraudtrucks.org/tentang-kami/
https://ojs.ejournalunigoro.org/
https://buslistrikmedan.id/area-cctv/
https://akashainternational.org/
https://kementeriankehutanan.com/
https://indonesiadanantara.org/
https://kementerianluarnegeri.com/
https://kementerianpertahanan.org/
https://revistas.unbosque.edu.co/
https://ucronias.unpaz.edu.ar/
https://sandiegohills.org/family-facilities/
https://jgp.ejournal.unri.ac.id/
https://jurnal.eka-prasetya.ac.id/
https://jurnal.isi-dps.ac.id/index.php/mudra
https://ejournalstisnuaceh.com/
https://ejss.eminentjournals.com/
https://jurnal.uisu.ac.id/index.php/languageliteracy
https://sep.ejournal.unri.ac.id/
https://jurnal.asrypersadaquality.com/
https://jurnal2.isi-dps.ac.id/index.php/amarasi
https://jurnal2.isi-dps.ac.id/index.php/retina
https://jurnal.uisu.ac.id/index.php/mesuisu
https://ejbm.eminentjournals.com/
https://plus.architecturalperiodicals.com/
https://journal.elsaonline.com/
https://publish.scholarsin.com/
https://negotiations.nbu.ac.in/
https://ojs.ejournalunigoro.com/index.php/sintesi
Điện thoại CSKH - Đặt lịch khám
19008082 - 0886.234.222Email hỗ trợ
bvhndkna.info@gmail.comKm5, Đại lộ Lê nin, phường Vinh Phú, tỉnh Nghệ An
02383.844.528
bvhndkna.info@gmail.com
19008082 - 0886.234.222
Copyright © 2026 BỆNH VIỆN HỮU NGHỊ ĐA KHOA NGHỆ AN