CHẤT LƯỢNG HÀNG ĐẦU - PHÁT TRIỂN CHUYÊN SÂU - NÂNG TẦM CAO MỚI
Thời gian làm việc: Khám bệnh: 7h-16h (Thứ 2-Thứ 6), 7h-12h (Sáng thứ 7), trừ nghỉ lễ ----- Tiếp nhận cấp cứu và điều trị nội trú 24/7, kể cả nghỉ lễ
CHẤT LƯỢNG HÀNG ĐẦU - PHÁT TRIỂN CHUYÊN SÂU - NÂNG TẦM CAO MỚI
Thời gian làm việc: Khám bệnh: 7h-16h (Thứ 2-Thứ 6), 7h-12h (Sáng thứ 7), trừ nghỉ lễ
Tiếp nhận cấp cứu và điều trị nội trú 24/7, kể cả nghỉ lễ
BỆNH VIỆN HỮU NGHỊ ĐA KHOA NGHỆ AN >
Đào tạo liên tục >
Bệnh Nội khoa >
Bệnh Nội tiết - Chuyển hoá >
U tuỷ thượng thận và u tế bào cận hạch thần kinh U tuỷ thượng thận và u tế bào cận hạch thần kinh
- U tuỷ thượng thận (Pheochromocytomas) và u tế bào cận hạch thần kinh (paragangliomas) là những khối u thần kinh nội tiết hiếm gặp phát sinh từ các tế bào cận hạch giao cảm và phó giao cảm, là những tế bào có nguồn gốc từ mào thần kinh có mặt trong tủy thượng thận và trong các mô dọc theo trục cạnh đốt sống.
- Pheochromocytoma đề cập đến một khối u thường tiết ra catecholamine trong tủy thượng thận.
- Paraganglioma đề cập đến các khối u ngoài tuyến thượng thận, có thể tiết ra hoặc không tiết ra catecholamine.
- Paragangliomas giao cảm là các khối u tiết catecholamine thường nằm trong phúc mạc (hoặc ít phổ biến hơn là xương chậu hoặc ngực).
- Paragangliomas phó giao cảm thường nằm ở đáy hộp sọ và cổ, và chỉ có khoảng 5% được báo cáo là tiết catecholamine.
- Hầu hết các pheochromocytomas và paragangliomas không phải là ác tính.
- Di căn được báo cáo ở:
- 5% -20% các bệnh nhân pheochromocytomas
- lên đến 35% ở paragangliomas giao cảm (nguy cơ thay đổi theo vị trí, với nguy cơ cao nhất liên quan đến paragangliomas trung thất và bụng)
- Các vị trí di căn phổ biến nhất bao gồm xương, phổi, gan và các hạch bạch huyết.
- Pheochromocytomas và paragangliomas có thể được phân loại theo tính chất gia đình (di truyền) hay lẻ tẻ.
- Khoảng 35% -40% của tất cả các pheochromocytomas và paragangliomas ở người lớn được báo cáo là có liên quan đến khuynh hướng di truyền.
- Lên đến 80% pheochromocytomas và paragangliomas ở trẻ em được báo cáo là có liên quan đến khuynh hướng di truyền.
- Pheochromocytomas gia đình có liên quan đến đột biến dòng tế bào mầm gây bệnh (đột biến di truyền xảy ra ở tất cả các tế bào của cơ thể) và có thể xảy ra ở những bệnh nhân mắc hội chứng ung thư di truyền, đa u tân sinh nội tiết loại 2, bệnh von Hippel-Lindau hoặc u xơ thần kinh loại 1.
- Hội chứng paraganglioma gia đình có liên quan đến đột biến trội tự phát được tìm thấy trong phức hợp succinate dehydrogenase (SDHD) II. Đột biến có thể xảy ra ở bất kỳ tiểu đơn vị gen nào (SDHA, SDHB, SDHC, SDHD) hoặc đồng yếu tố, tuy nhiên, đột biến mất chức năng trong tiểu đơn vị SDHB thường liên quan nhất đến pheochromocytomas.
- Pheochromocytomas lẻ tẻ và paragangliomas không có mối liên hệ di truyền được biết đến; tuy nhiên, tỷ lệ bệnh nhân gia tăng (lên đến 40% vào năm 2019) được báo cáo là có đột biến mầm bệnh, ngay cả khi không có tiền sử gia đình hoặc cá nhân liên quan.
- Các triệu chứng và dấu hiệu là do tăng tiết catecholamine hoặc hiệu ứng khối (mass).
- Tam chứng cổ điển bao gồm đau đầu kịch phát, đánh trống ngực và đổ mồ hôi nhiều.
- Các dấu hiệu cổ điển khác của sự dư thừa catecholamine bao gồm:
- tăng huyết áp (có thể kéo dài, kịch phát hoặc tư thế đứng)
- Sợ hãi hoặc lo lắng
- rối loạn nhịp tim
- Các dấu hiệu và triệu chứng không phải cổ điển có thể bao gồm:
- xanh xao
- buồn nôn hoặc nôn
- giảm cân
- sốt
- Mệt mỏi
- tăng đường huyết
- Biểu hiện lâm sàng có thể phản ánh sự bài tiết tương đối của norepinephrine so với epinephrine bởi khối u.
- Các khối u chủ yếu tiết ra norepinephrine thường liên quan đến tăng huyết áp kéo dài.
- Các khối u chủ yếu tiết ra epinephrine (khối u trong tuyến thượng thận) thường biểu hiện với tăng huyết áp kịch phát và các đợt hạ huyết áp thế đứng do tác dụng epinephrine tại thụ thể adrenergic beta 2.
- Pheochromocytomas và paragangliomas không được điều trị có liên quan đến tỷ lệ mắc bệnh tim mạch và tỷ lệ tử vong cao.
Chẩn đoán
- Pheochromocytomas không triệu chứng hoặc paragangliomas (PPGL) thường được chẩn đoán ngẫu nhiên ở những bệnh nhân:
- một khối tuyến thượng thận hoặc sau phúc mạc trên hình ảnh được thực hiện vì những lý do không liên quan
- kết quả xét nghiệm sinh hóa hoặc hình ảnh bất thường trong quá trình theo dõi một người có nguy cơ di truyền hoặc trong quá trình sàng lọc một thành viên gia đình có nguy cơ
- Nghi ngờ pheochromocytoma hoặc paraganglioma ở những bệnh nhân có biểu hiện lâm sàng nhất quán cộng với bất kỳ điều nào sau đây:
- tăng huyết áp dễ bay hơi, hoặc kháng trị liệu
- phản ứng huyết áp nghịch lý trong khi phẫu thuật hoặc gây mê
- Incidentaloma tuyến thượng thận (incidentaloma là một khối u thượng thận không có triệu chứng lâm sàng được phát hiện một cách tình cờ trong quá trình thăm dò hình ảnh cho một bệnh lý khác)
- Cơn hoảng sợ đột ngột
- tiền sử gia đình mắc hội chứng di truyền liên quan đến pheochromocytoma hoặc paraganglioma
- Các loại thuốc được biết là dẫn đến khủng hoảng hoặc gây ra các tác dụng phụ ở những người bị pheochromocytoma hoặc paraganglioma
- Chẩn đoán xác định pheochromocytoma hoặc paraganglioma giao cảm đòi hỏi bằng chứng sinh hóa về sự giải phóng quá mức catecholamine và định vị giải phẫu của khối u.
- Đo metanephrine trong huyết tương và nước tiểu (Khuyến cáo mạnh). Ở khoảng 80% bệnh nhân bị pheochromocytoma, metanephrine huyết tương được báo cáo là tăng cao hơn 4 lần giới hạn trên của mức bình thường.
- Cân nhắc xét nghiệm ức chế clonidine để phân biệt các xét nghiệm metanephrine huyết tương dương tính giả và dương tính thật, hoặc trong việc đánh giá bệnh nhân có nồng độ catecholamine và metanephrine ranh giới.
- Thực hiện các thăm dò hình ảnh để định vị khối u sau khi chẩn đoán sinh hóa được thiết lập (Khuyến nghị mạnh mẽ).
- Định vị khối u là cần thiết vì những khối u này không thể được phân biệt bởi các đặc điểm mô học.
- Thực hiện chụp cắt lớp vi tính bụng (CT) hoặc chụp cộng hưởng từ (MRI).
- Nếu hình ảnh bụng âm tính, hãy xem xét chụp MRI đáy sọ, cổ, ngực và xương chậu.
- Do tăng nguy cơ mắc các khối u bổ sung hoặc bệnh di căn, nếu khối u kích thước lớn (ví dụ: đường kính > 6 cm) hoặc ngoài tuyến thượng thận, đa ổ (trừ nền sọ và paragangliomas cổ), hoặc tái phát, hãy cân nhắc thực hiện chụp ảnh chức năng bằng bất kỳ cách nào sau đây:
- F-fluorodeoxyglucose (F-FDG)-PET-CT
- xạ hình dán nhãn I metaiodobenzylguanidine (I-MIBG)
- Ga-DOTATATE-PET-CT ·
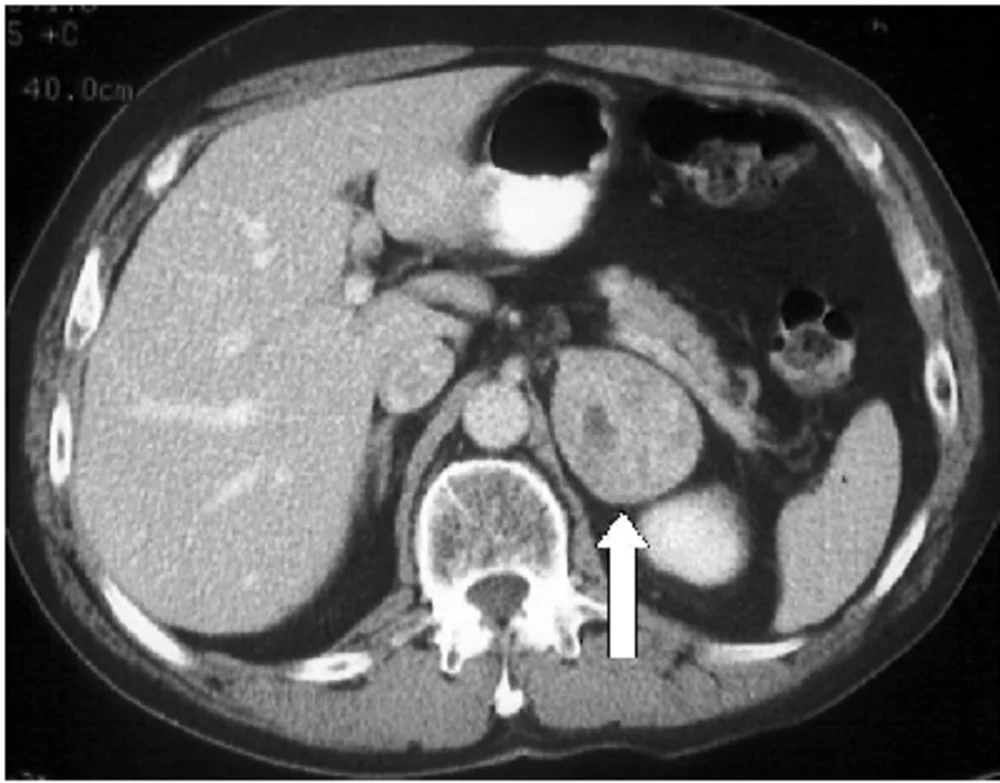
Hình 1. Pheochromocytoma tuyến thượng thận trái Biểu hiện không đồng nhất 5 cm (2 inch), tăng độ tương phản, pheochromocytoma tuyến thượng thận trái (mũi tên) trên CT tuyến thượng thận tăng cường độ tương phản. Viết tắt: CT, chụp cắt lớp vi tính.
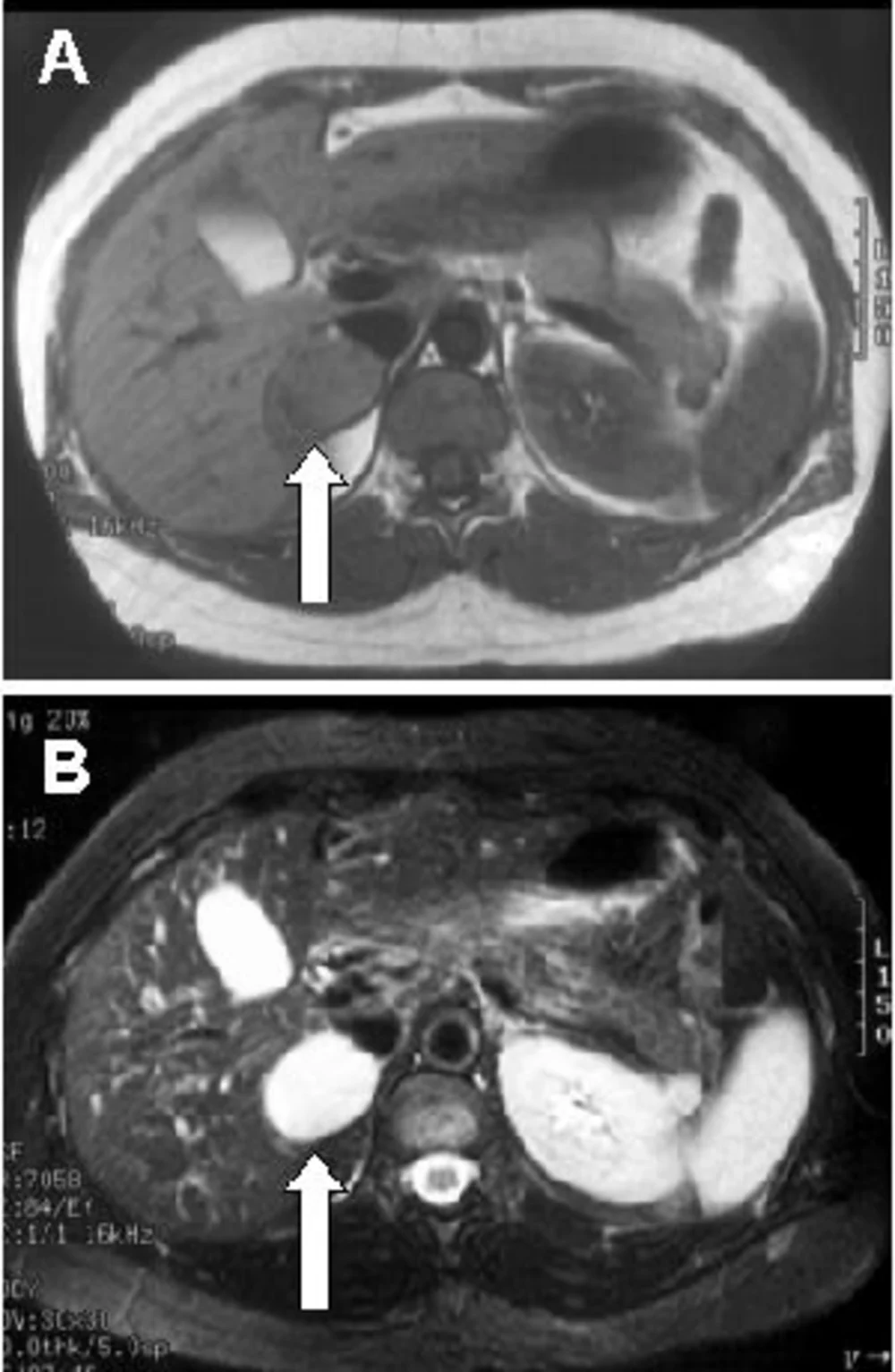
Hình 2. Hình ảnh cộng hưởng từ tuyến thượng thận
A) Hình ảnh có trọng số T1 cho thấy khối u 6 cm (2,4 inch) phát sinh từ tuyến thượng thận phải (mũi tên). B) Hình ảnh trọng số T2 cho thấy cường độ tín hiệu cao phù hợp với pheochromocytoma (mũi tên). Khi cắt bỏ tuyến thượng thận nội soi, một pheochromocytoma 6- x 5- x 4,5 cm (2,4- x 2- x 1,8 inch) đã được loại bỏ.
- Sau khi chẩn đoán được thiết lập, hãy thực hiện xét nghiệm di truyền để phát hiện đột biến đặc hiệu nhằm hướng dẫn điều trị và theo dõi tiếp theo.
- Chẩn đoán bệnh di căn dựa trên bằng chứng về khối u ở những vị trí mà tế bào chromaffin thường không xuất hiện (không có xét nghiệm giải phẫu bệnh lý đáng tin cậy nào có thể phân biệt giữa khối u nguyên phát không di căn và di căn).
Điều trị
- Trước khi điều trị dứt điểm bằng phẫu thuật, cần quản lý nội khoa trước phẫu thuật để kiểm soát các triệu chứng do catecholamine gây ra, ổn định huyết áp và nhịp tim của bệnh nhân, đồng thời ngăn ngừa các biến chứng đe dọa tính mạng (bao gồm cả cơn tăng huyết áp) trong quá trình phẫu thuật.
- Bắt đầu điều trị nội khoa trước phẫu thuật ít nhất 7-14 ngày trước khi phẫu thuật để có đủ thời gian cho huyết áp và nhịp tim bình thường hóa (Khuyến nghị mạnh mẽ).
- Khuyến cáo phong tỏa adrenergic trước phẫu thuật ở tất cả các bệnh nhân có pheochromocytomas chức năng nội tiết tố và paragangliomas vì việc giải phóng nồng độ catecholamine lưu hành cao trong quá trình phẫu thuật có thể gây ra cơn tăng huyết áp và rối loạn nhịp tim (Khuyến cáo mạnh).
- Thuốc chẹn alpha được sử dụng để quản lý ban đầu (Khuyến nghị yếu).
- Phenoxybenzamine là một chất đối kháng alpha không chọn lọc, không thể đảo ngược, và thường được sử dụng như là liệu pháp ban đầu do tác dụng không thể đảo ngược và kéo dài của nó; tuy nhiên, việc sử dụng nó có thể bị giới hạn bởi tính khả dụng, tác dụng phụ và chi phí.
- Các lựa chọn thay thế cho phenoxybenzamine bao gồm các thuốc đối kháng alpha-1-adrenergic chọn lọc như prazosin, doxazosin hoặc terazosin.
- Thuốc chẹn beta có thể được sử dụng để kiểm soát nhịp tim nhanh chỉ sau khi kiểm soát huyết áp thành công bằng thuốc chẹn alpha. Thuốc chẹn beta không nên được sử dụng trong trường hợp không có thuốc chẹn alpha do sự kích thích không bị cản trở của các thụ thể alpha adrenergic dẫn đến cơn tăng huyết áp.
- Cân nhắc thuốc chẹn kênh canxi ở những bệnh nhân không kiểm soát huyết áp không đầy đủ chỉ bằng thuốc chẹn alpha, hoặc thay thế cho thuốc chẹn alpha ở những bệnh nhân có tác dụng phụ nghiêm trọng. Đơn trị liệu với thuốc chẹn kênh canxi cũng có thể được xem xét ở những bệnh nhân có huyết áp bình thường hoặc tăng huyết áp trước phẫu thuật rất nhẹ.
- Trong khi các mục tiêu tim mạch tối ưu vẫn chưa được xác định, các mục tiêu sau đây được coi là hợp lý:
- huyết áp mục tiêu < 130/80 mm Hg ở tư thế nằm ngửa và huyết áp tâm thu > 90 mm Hg ở tư thế đứng
- nhịp tim mục tiêu là 60-70 nhịp mỗi phút khi ngồi và 70-80 nhịp mỗi phút khi đứng
- Nên bắt đầu chế độ ăn nhiều natri (ví dụ 5.000 mg/ngày) với uống nhiều nước (ví dụ 2,5 L/ngày) kết hợp với phong tỏa adrenergic trước khi phẫu thuật (thường bắt đầu khoảng 7-14 ngày trước khi phẫu thuật).
- Phẫu thuật là phương pháp điều trị được lựa chọn ở những bệnh nhân mắc bệnh không di căn hoặc di căn có thể cắt bỏ.
- Các lựa chọn phẫu thuật bao gồm cắt bỏ toàn bộ tuyến thượng thận hoặc cắt bỏ tuyến thượng thận bảo tồn phần vỏ (cắt bỏ một phần tuyến thượng thận), cả hai đều có thể được thực hiện thông qua phương pháp nội soi hoặc mở.
- Khuyến cáo phẫu thuật cắt bỏ tuyến thượng thận xâm lấn tối thiểu (chẳng hạn như cắt bỏ tuyến thượng thận nội soi) cho hầu hết các pheochromocytomas (Khuyến cáo mạnh).
- Phẫu thuật mở có thể cần thiết ở những bệnh nhân có pheochromocytomas kích thước lớn (ví dụ, > 6 cm) hoặc xâm lấn.
- Phẫu thuật mở thường được thực hiện đối với paragangliomas do tăng khả năng di căn và vị trí ở những khu vực khó tiếp cận bằng nội soi, nhưng phẫu thuật nội soi là một lựa chọn cho các paragangliomas nhỏ, không xâm lấn ở những vị trí dễ tiếp cận.
- Quản lý ở trẻ em
- Tương tự như người lớn, điều trị ở trẻ em bao gồm chuẩn bị trước phẫu thuật bằng thuốc chẹn alpha, thuốc chẹn beta và các loại thuốc khác để kiểm soát tăng huyết áp, rối loạn nhịp tim và tăng thể tích, đồng thời ngăn ngừa các biến chứng của cơn tăng huyết áp trước khi điều trị dứt điểm bằng phẫu thuật.
- Giảm huyết áp mục tiêu là < phân vị thứ 50 cho tuổi và chiều cao.
- Cắt bỏ qua nội soi ổ bụng và các thủ thuật bảo tồn vỏ thượng thận (cắt bỏ một phần tuyến thượng thận) thường được ưu tiên ở trẻ em, đặc biệt là ở trẻ em mắc bệnh thượng thận hai bên.
- Cung cấp dịch vụ chăm sóc theo dõi suốt đời với một nhóm đa ngành bao gồm bác sĩ nội tiết giàu kinh nghiệm, bác sĩ thận, bác sĩ phẫu thuật, cố vấn di truyền, bác sĩ ung thư và bác sĩ X quang / chuyên gia y học hạt nhân.
- Lên đến 80% pheochromocytomas và paragangliomas ở trẻ em được báo cáo là có liên quan đến khuynh hướng di truyền.
- Tất cả trẻ em bị đột biến cần được theo dõi chặt chẽ do khả năng tái phát và di căn.
- Quản lý ở phụ nữ mang thai
- Quản lý nội khoa trước phẫu thuật là cần thiết để giảm thiểu nguy cơ biến chứng nghiêm trọng do giải phóng lớn catecholamine.
- Thuốc chẹn alpha như phenoxybenzamine và doxazosin có thể đi qua nhau thai và được chuyển vào sữa mẹ, do đó nên theo dõi trẻ sơ sinh trong 3 ngày đầu sau sinh.
- Xem xét huyết áp mục tiêu là 140/90 mm Hg.
- Quản lý phẫu thuật thay đổi tùy theo thời gian mang thai khi khối u được phát hiện.
- Cân nhắc cắt bỏ khối u bằng nội soi trước 24 tuần tuổi thai vì nguy cơ sảy thai tự nhiên là thấp nhất (ưu tiên phương pháp tiếp cận qua phúc mạc với bệnh nhân ở tư thế nằm nghiêng để cắt bỏ khối u bằng nội soi).
- Trì hoãn việc cắt bỏ khối u cho đến khi hoặc sau khi sinh nếu bệnh nhân được chẩn đoán và phát hiện trong tam cá nguyệt thứ ba, và xem xét điều trị nội khoa để kiểm soát lượng catecholamine dư thừa cho đến lúc đó.
- Sinh mổ là phương pháp ưu tiên để sinh nở an toàn.
- Các chiến lược bao gồm kết hợp sinh mổ và cắt bỏ khối u trong cùng một cuộc phẫu thuật, hoặc hoãn phẫu thuật trong vài tuần sau khi sinh.
- Điều trị pheochromocytomas và paragangliomas di căn
- Cung cấp dịch vụ chăm sóc hỗ trợ trước phẫu thuật để làm giảm các triệu chứng liên quan đến dư thừa catecholamine và giảm nguy cơ cơn tăng huyết áp quanh phẫu thuật.
- Xem xét cắt bỏ giảm số lượng tế bào để giảm gánh nặng khối u và kiểm soát sự tăng tiết nội tiết tố (Khuyến cáo yếu).
- Xạ trị với các chất mang nhãn phóng xạ được nhắm mục tiêu, chẳng hạn như iốt 131-metaiodobenzylguanidine (131I-MIBG) là một lựa chọn cho bệnh nhân có gánh nặng khối u cao hoặc bệnh không thể cắt bỏ với sự hấp thu tốt 123I-MIBG.
- Theo dõi lâu dài trong ≥ 10 năm sau khi phẫu thuật thành công pheochromocytoma / paraganglioma không di căn.
- Cân nhắc đo nồng độ metanephrine trong huyết tương hoặc nước tiểu khi theo dõi để xác định bệnh dai dẳng (Khuyến cáo yếu).
- Cân nhắc xét nghiệm sinh hóa suốt đời hàng năm để xác định bệnh di căn hoặc tái phát (Khuyến cáo yếu).
Chủ đề liên quan
- Đa u tân sinh nội tiết loại 2A
- Đa u tân sinh nội tiết loại 2B
- U sợi thần kinh Loại 1
- Bệnh Von Hippel-Lindau
Bs.Ths. Lê Đình Sáng (Dịch và tổng hợp từ Dynamed)
TÀI LIỆU THAM KHẢO
Hướng dẫn (GUIDELINES)
Hướng dẫn của Hoa Kỳ
- Hướng dẫn của Mạng lưới Ung thư Toàn diện Quốc gia (NCCN) về các khối u nội tiết thần kinh có thể được tìm thấy tại trang web NCCN (yêu cầu đăng ký miễn phí)
- Hướng dẫn thực hành lâm sàng của Hiệp hội Nội tiết về pheochromocytoma và paraganglioma có thể được tìm thấy trong J Clin Endocrinol Metab 2014 Tháng sáu;99(6):1915
- Hướng dẫn của Hiệp hội bác sĩ phẫu thuật nội soi và đường tiêu hóa Hoa Kỳ (SAGES) về điều trị xâm lấn tối thiểu bệnh lý tuyến thượng thận có thể được tìm thấy tại SAGES 2013 Feb
Hướng dẫn của Canada
- Báo cáo thực hành tốt nhất của Hiệp hội Tiết niệu Canada (CUA) để theo dõi lâu dài sau khi cắt bỏ pheochromocytoma có thể được tìm thấy trong Can Urol Assoc J 2019 Dec;13(12):372full-text
- Hướng dẫn thực hành lâm sàng của Dịch vụ Y tế Alberta (AHS) về các chất tương tự somatostatin để quản lý khối u nội tiết thần kinh có thể được tìm thấy tại AHS 2015 Mar PDF
- Hướng dẫn dựa trên bằng chứng về chăm sóc ung thư Ontario (CCO) về liệu pháp hạt nhân phóng xạ cho các khối u ác tính nội tiết thần kinh có thể được tìm thấy tại CCO 2011 Aug 15
Hướng dẫn Châu Âu
- Hướng dẫn thực hành lâm sàng của Hiệp hội Ung thư Y tế Châu Âu (ESMO) về chẩn đoán, điều trị và theo dõi ung thư biểu mô adrenocortical và phaeochromocytomas ác tính có thể được tìm thấy trong Ann Oncol 2020 Nov;31(11):1476
- Hướng dẫn của Hiệp hội Nội tiết Châu Âu (ESE) về
- Hướng dẫn của Hiệp hội Y học Hạt nhân Châu Âu (EANM) 2012 về hình ảnh hạt nhân phóng xạ của phaeochromocytoma và paraganglioma có thể được tìm thấy trong Eur J Nucl Med Mol Hình ảnh 2012 Tháng mười hai;39(12):1977toàn văn
- Hướng dẫn đồng thuận của Hiệp hội Khối u Nội tiết Thần kinh Châu Âu (ENETS) về quản lý bệnh nhân có khối u thần kinh tiêu hóa có thể được tìm thấy trong Nội tiết thần kinh 2016;103(2):117
- Khuyến nghị của Hiệp hội Quản lý Nội tiết Ba Lan về bệnh viêm tuyến thượng thận ở người lớn có thể được tìm thấy trong Endokrynol Pol 2016;67(2):234
- Hướng dẫn lâm sàng của Hiệp hội Ung thư Y tế Tây Ban Nha (Sociedad Española de Oncología Médica [SEOM]) về chẩn đoán và điều trị các khối u thần kinh nội tiết dạ dày ruột (GEP NENs) có thể được tìm thấy trong Clin Transl Oncol 2019 Jan;21(1):55full-text
TÀI LIỆU THAM KHẢO
- Neumann HPH, Young WF Jr, Eng C. Pheochromocytoma and Paraganglioma. N Engl J Med. 2019 Aug 8;381(6):552-65, commentary can be found in N Engl J Med 2019 Nov 7;381(19):1882
- Else T, Greenberg S, Fishbein L. Hereditary Paraganglioma-Pheochromocytoma Syndromes. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews [Internet]. Seattle, WA: University of Washington, Seattle; 2018 Oct 4.
- Gunawardane PTK, Grossman A. Phaeochromocytoma and Paraganglioma. Adv Exp Med Biol. 2017;956:239-59
- Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42
- Fassnacht M, Assie G, Baudin E, et al. European Society for Medical Oncology (ESMO) Guidelines Committee. Adrenocortical carcinomas and malignant phaeochromocytomas: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2020 Aug 24 early online
- Jasim S, Jimenez C. Metastatic pheochromocytoma and paraganglioma: Management of endocrine manifestations, surgery and ablative procedures, and systemic therapies. Best Pract Res Clin Endocrinol Metab. 2020 Mar;34(2):101354

CHẤT LƯỢNG HÀNG ĐẦU - PHÁT TRIỂN CHUYÊN SÂU - NÂNG TẦM CAO MỚI
Địa chỉ
Km5, Đại lộ Lê nin, phường Vinh Phú, tỉnh Nghệ An
Điện thoại
02383.844.528
Email
bvhndkna.info@gmail.com
Điện thoại CSKH - Đặt lịch khám
19008082 - 0886.234.222
Thời gian làm việc: Khám bệnh: 7h-16h (Thứ 2-Thứ 6), 7h-12h (Sáng thứ 7), trừ nghỉ lễ ----- Tiếp nhận cấp cứu và điều trị nội trú 24/7, kể cả nghỉ lễ
Copyright © 2026 BỆNH VIỆN HỮU NGHỊ ĐA KHOA NGHỆ AN
